ͨ������^�ȼ��Ą��ּ����Pע�tˎ��ˎƷ�ИI����^�˜ʣ���ˎƷ���a�|������Ҏ����2010����ӆ����(�°�GMP)��2011��3��1����ʩ�У�������Ҫ��B�°�GMP���P�ڝ����ȵȼ��е�A��B��C��D�Ă����e���Լ��°�GMP�c98�����P�ڝ����ȵȼ��ą^�e��
GMP��A��B��C��D���������Є��o�B֮�֣����ټ����f����ʮ�f���t�����o���o�B֮�֣�����֮�g�������@�IJ���°�GMP����ISO14644��Ҏ�����w�˜����£��°�GMP�����˚W�˺�����WHO��A��B��C��D�ּ��˜ʣ������o��ˎƷ���a�ĝ����ȼ��e����˷dz����w��Ҫ��
�o�B�y������ָ�����O����Ѱ��b�;w����δ�\���қ]�в����ˆT�ڬF���Ġ�B��
�ӑB�y������ָ�����O������A���Ĺ�ˇģʽ�\������Ҏ�������IJ����ˆT�ڬF�������Ġ�B��
���°�GMP�˜��У�A�^�ĄӑB���o�B�Լ�B�^���o�B��Ҫ���100�������京�x��ͬ��A�^��100���������������Ҫ��B�^��100���t�o��Ҫ���°�GMPA�^�c98��GMP�е�100�����ƣ�B�^�c98��GMP�е����PҎ�����ܴ����֞��o�B�ټ��̈́ӑB�f�������҇���GMPֻҪ��100����1�f���ı����^��ȡ����ڰټ��^(A�^)�����������٣��°�GMP�Ę˜���0.45±20����98��GMP��Ҏ����0.2��0.5m/s�����g������ͬ,�°�GMPҪ��������ͬ���g���e�ĉ���鲻����10Pa����98��GMPҪ����5Pa����������ıO�غ�ȡ�Ә˜�Ҳ�в�ͬ��
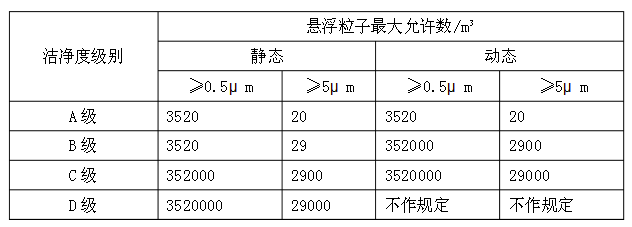
ע��1����_�JA������^�ļ��e��ÿ���ɘ��c�IJɘ�����������1�����ס�A������^�՚�Ҹ����ӵļ��e��ISO4.8����≥5.0μm�đҸ����Ӟ��Ș˜ʡ�B������^���o�B���Ŀ՚�Ҹ����ӵļ��e��ISO5��ͬ�r�������ЃɷN�����đҸ����ӡ�����C������^���o�B�̈́ӑB�����ԣ��՚�Ҹ����ӵļ��e�քe��ISO7��ISO8������D������^���o�B���՚�Ҹ����ӵļ��e��ISO8���yԇ�����Ʌ���ISO14644-1��
2���ڴ_�J���e�r������ʹ�òɘӹ��^�̵ı�yʽ�m������Ӌ����������≥5.0μm�Ҹ��������h�̲ɘ�ϵ�y���L�ɘӹ��г������چ�����ϵ�y�У��������õȄ����W��ȡ���^��
3���ӑB�yԇ���ڳ�Ҏ���������B��ģ�M���b�^�����M�У��C���_���ӑB�ĝ����ȼ��e�������B��ģ�M���bԇ�Ҫ����“����r”���M�ЄӑB�yԇ��
d������^
����D������^��ˇ�l�������ƶȼ����I����ͷ������Ա��C����Ĺ�ˇ���E�����_�؈��С�D������^���a�^���е����ϣ����a���̼��O����l��������D������^�����_��һ�����a�^��ˇ�l����ȫ��Ҫ���⣬߀����Mһ���ڃ��������_������Ҫ��
1��ԭ�o���l�����M��D������^��ԭ�o�ϡ��Ȱ��b���ϡ����������߾����ھ��_�g������ң���������M���坍����ȥ��Ⱦ����Ƥ����Q�ɝ����IJ��P�Ͱ�����w�������ý���75%�Ҵ���Һ��Ĩ���������������������15��犣�ͨ�^���f������l���M��D������^���M�띍��^�ȵIJ��ϑ�����������ȣ������������M��������^�Ȳ��ܴ�Ŵ�����������ϼ��c���a�o�P�����ϡ�����^�ȵ�ԭ�o���ϣ��Ȱ��b���ϡ����������߱�횷��ڲ�Ӱ푻�����Ӱ푚�����Ҏ��λ�á����ϝ�������

2�����a�^���е��l��������^���坍һ�����ڹ�������Y�����M�У������Ҫ���������aǰ�ٴ��M���坍���������a����ڃ������{ϵ�y�_�C�\���_���ԃ��r�g�Ժ��_ʼ�M�С����a�_ʼǰ,���������������_�桢���ߣ��cˎ����|���O����漰�㲿���M��һ������̎��, ���a�Y����,��횇������D������^�坍Ҏ�̡����a�^�����ϑ��ѷ���ָ����λ�ã��U����Ҫ�ռ���ָ���ļ��w�������ȣ����ڹ����Y���r������ȥ�����QƷ�N�����չ����Y����회������桢�ذ塢�������_�桢���߰������坍Ҏ���坍��������
3���O���l��������^�O���l�����_��һ�����a�^��ˇ�l��Ҏ��Ҫ����,߀��_������Ҫ���f�������l�ң��ǝ���^�cһ�����a�^�ĸ����O�䡢�Á���ֹ�ǝ����՚⌦�����՚����Ⱦ����ˣ����f�������l�ң����T���i������ͬ�r�_������������·��ֱ�ӽ��|ˎƷ���O���㲿�����ڸ��QƷ�NҎ������r,��횲�����ϴ������̎�����ֲ������OʩҪ��Ҏ���Ę˜ʲ���Ҏ���M�б��B���坍���Q���y���K������ÿ�α���ڹ�ˇ����ǰ30��犆��ӡ��O��ʹ�õĝ���������s�������cˎƷ���������Ȱ��b���ϵȽ��|��������Ҫ�����IJ�λ�M�������c�O��ͮaƷ���|���_��̎����|��ָ��_���O��ľS�ޱ����ϵ�yͣ�a����r���M�У��S�Y�������r�����F�������坍Ҏ�̌�ȫϵ�y�M�Џص��坍���������ܵ���Ҏ���M�И��������������